Der Rollstuhl wird Sophia Elena wahrscheinlich erspart bleiben
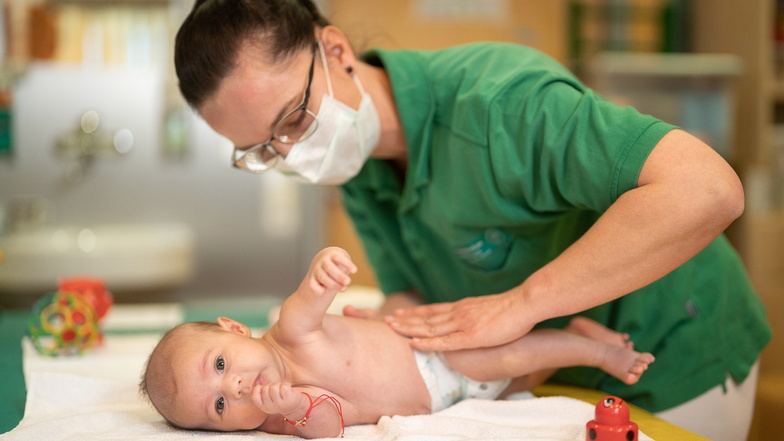
Das kleine Mädchen ist nicht begeistert, als sie von Ilka Lehnert auf den Bauch gedreht wird. Die Kinderphysiotherapeutin am Uniklinikum Dresden untersucht auf diese Weise, ob das Mädchen schon allein ihren Kopf halten kann. Dass sie das schafft, ist für ihre Eltern ein Wunder. Denn Sophia Elena leidet an Spinaler Muskelatrophie – SMA.
In diesem Text lesen Sie:
- Baby John aus Sebnitz bekam als erstes Kind in Sachsen ein neues gentherapeutisches Medikament gegen SMA – wie es ihm heute geht.
- Welche Zeichen bei Babys auf SMA hinweisen können.
- Wie viele Kinder in Dresden wegen SMA bereits behandelt werden.
- Ob das Medikament auch Komplikationen haben kann.
Etwa eines von 8.000 Neugeborenen kommt in Deutschland mit dieser Erkrankung zur Welt. Darunter Baby John aus Sebnitz, dessen Schicksal vor zweieinhalb Jahren viele Menschen bewegt hat. Aufgrund eines Gendefekts werden die motorischen Nerven nicht mehr mit einem notwendigen Protein versorgt und sterben ab. Folgen sind eine fortschreitende Muskelschwäche und Lähmungen, die auch die Atemmuskulatur betreffen. Unbehandelt erreichen Kinder mit der schwersten Form der Erkrankung selten das zweite Lebensjahr. Doch es gibt Behandlungsmöglichkeiten. Je früher sie einsetzen – am besten, bevor erste Symptome auftreten –, umso weniger lebenswichtige Fähigkeiten gehen verloren.
Zwei Kinder – Sophia Elena und Peter – bekamen in diesem Jahr am Uniklinikum Dresden kurze Zeit nach ihrer Geburt ein hochwirksames Medikament. Bei ihnen war der Gendefekt bereits im Rahmen des Baby-Screenings entdeckt worden. Seit Oktober 2021 werden Neugeborene bundesweit routinemäßig darauf untersucht. Die Elterninitiative "Forschung und Therapie für SMA" hatte viele Jahre darum gekämpft.
Teuerstes Medikament
Die Infusion mit dem Medikament Zolgensma ist eine Genersatztherapie. Mit nur einer Behandlung wird das defekte Gen ausgetauscht. Eine sehr teure Therapie – allein das Medikament kostet rund zwei Millionen Euro, hinzu kommen die Behandlungskosten und die jahrelange Nachsorge im spezialisierten Zentrum. Deshalb sind an die Therapie strenge Forderungen geknüpft. In Ostdeutschland haben nur zwei Kinderkliniken – das Uniklinikum Dresden und die Charité Berlin – diese Berechtigung. In Dresden werden derzeit 32 Kinder und Jugendliche mit dieser neuromuskulären Erkrankung behandelt, zehn davon erhalten Zolgensma.
"Peter und Sophia Elena haben die Gabe des Medikaments problemlos vertragen, sodass sie bereits entlassen werden konnten. Beide Säuglinge kommen in den nächsten Monaten nun wöchentlich zu ambulanten Nachsorgekontrollen in die Klinik", sagt Professorin Maja von der Hagen, Leiterin der Abteilung Neuropädiatrie an der Uni-Kinderklinik. Die Daten der Kinder fließen in ein nationales Patientenregister und in die weitere SMA-Forschung ein.
Kinderphysiotherapeutin Ilka Lehnert ist mit dem Ergebnis zufrieden: "Sophia Elena kann die angehockten Beinchen schon gegen die Schwerkraft oben halten. Sie fallen nicht gleich wieder in die Froschhaltung auf die Unterlage zurück, wie es bei der SMA sonst typisch ist. Auch die Gelenke zeigen keine Blockierungen – die Kleine ist ein ganz normal entwickeltes Kind."
Sophia Elenas Schwester Antonia hatte nicht so viel Glück. Die heute Fünfjährige hat erst im Alter von 15 Monaten ein Medikament gegen SMA bekommen, nachdem sie bereits erste selbstständige Schritte gemacht hatte. "Wir sind aus Rumänien nach Deutschland gekommen, weil es hier eine Behandlung für unser Kind gab", sagt Claudiu Radu, der Vater der beiden Mädchen. Von ihrem Wohnort Plauen aus fahren sie dazu mit Antonia regelmäßig in die Uni-Kinderklinik Jena. Denn Antonia bekam Spinraza – ein weiteres gegen SMA zugelassenes Medikament. Im Gegensatz zu Zolgensma muss es etwa alle vier Monate in den Rückenmarkskanal verabreicht werden. "Es ersetzt das defekte Gen nicht, sondern aktiviert ein Ersatzgen, das dann in Protein umgesetzt wird", sagt Kinderneurologe Dr. Martin Smitka. Schon bald kann sie das Medikament als Saft erhalten. "Es hat sich in den letzten fünf Jahren viel in der SMA-Therapie getan", so Smitka.

"Wir waren voller Schmerz"
Laufen kann Antonia nicht. Sie sitzt in ihrem pinkfarbenen Kinderrollstuhl, spielt am Tablet und schaut ihrer Schwester bei der Untersuchung zu. "Sie ist ein fröhliches Mädchen, geht in den Kindergarten und bald in eine Schule für Körperbehinderte nach Chemnitz. Nicht auszudenken, wie es ihr ohne diese Behandlung ergehen würde", so Claudiu Radu. Antonia lerne jeden Tag etwas Neues. "Wir haben einige Reha-Sportgeräte zu Hause, da macht sie gute Fortschritte.
"Als bei Sophia Elena im Neugeborenen-Screening auch diese Krankheit entdeckt wurde, waren wir voller Schmerz. Jetzt schauen wir positiver nach vorn und haben Hoffnung, dass die Therapie gut anschlägt", so der Vater.
"Die uns vorliegenden wissenschaftlichen Daten bestärken uns in der Hoffnung, dass die beiden Neugeborenen dank dieser frühen Gabe des Medikaments keine oder nur geringe Symptome entwickeln werden", sagt Professorin von der Hagen. Für Martin Smitka ist es eine Frage der Zeit: "Wenn wir die Kinder im Neugeborenen-Screening diagnostizieren und behandeln können, bevor sie Symptome entwickeln, werden wir bald gar keine Kinder mehr mit schweren SMA-Symptomen sehen – ein großes Glück."
Lebensfroh trotz Handicap
Baby John aus Sebnitz war das erste Kind in Sachsen, das mit Zolgensma behandelt werden konnte. Doch vor zweieinhalb Jahren durfte die Krankenkasse die Behandlungskosten noch nicht übernehmen, da das Medikament nicht zugelassen war. Auch Johns Eltern haben gekämpft und gewonnen. Jetzt ist John drei Jahre alt. "Er bekommt bald seinen Kinderrollstuhl", sagt seine Mutter. Auch wenn er vielleicht nie laufen können wird, sei er ein lebensfrohes Kind. Mutter und Sohn waren kürzlich das erste Mal gemeinsam zur Reha. "Das haben wir sehr genossen", so die Mutter.
Etwa jeder 50. Mensch ist Martin Smitka zufolge Träger des SMA-Gens. Damit die Krankheit zum Ausbruch kommt, müssen beide Eltern Genträger sein. "Dass wir beide diese Genveränderung haben, wussten wir nicht. Niemand in unserer Familie hatte bisher diese Krankheit", so Claudiu Radu. Mittlerweile kann die Erkrankung auch schon bei Schwangeren durch die Untersuchung von Fruchtwasser erkannt werden. "Das haben wir nicht gemacht. Für uns wäre es auch nie infrage gekommen, uns gegen unser Kind zu entscheiden", sagt Sophia Elenas Vater.
Der Gabe des Gen-Therapeutikums waren aufwendige Untersuchungen vorausgegangen. Das modifizierte Gen wird mithilfe eines Vektors – eines abgeschwächten Adenovirus – in den Körper eingeschleust. "Als Vektor werden solche Viren ausgewählt, die die entsprechende Körperregion im Falle einer Infektion befallen", erklärt Dr. Smitka. Vorher müsse sichergestellt werden, dass der Patient keine Antikörper gegen dieses Virus hat.
Neues Gen nicht vererbt
Auch eine gute Leber- und Herzgesundheit seien wichtige Voraussetzungen. Sonst könnte das Medikament schwerwiegende Reaktionen im Organismus auslösen oder unwirksam sein. Die Infusion wird den Kindern auf der pädiatrischen Intensivstation verabreicht. So können die Säuglinge während und nach der Gabe des Genmedikaments engmaschig überwacht werden. "Das ersetzte Gen wird nicht in die DNA eingebaut, sondern in den Zellkern", so Dr. Smitka. Damit könne es nicht vererbt werden. Spätere Nachkommen von SMA-Patienten müssten also ebenfalls behandelt werden, wenn bei ihnen diese Krankheit festgestellt wird. Doch daran denken die Eltern von Sophia Elena und Antonia noch nicht. Sie sind sehr dankbar, dass ihren Kindern mit modernen Medikamenten geholfen wurde und ihnen noch hoffentlich viel glückliche Lebenszeit bevorsteht.
Vom Test zur SMA-Therapie in drei Wochen
- Lebenstag 3–5: Für das Neugeborenen-Screening wird allen Babys etwas Blut aus der Ferse entnommen und auf eine Filterkarte getropft. Untersucht wird es derzeit auf 19 Krankheiten, seit Oktober auch auf SMA.
- Lebenstag 5–10: Ist der Test auf Spinale Muskelatrophie positiv, informiert der Screeningarzt sofort das zuständige neuromuskuläre Zentrum und übermittelt die Kontaktdaten der Eltern. Der Kinderneurologe des Zentrums bittet die Eltern zum Aufklärungsgespräch über die therapeutischen Möglichkeiten. Beim Kind erfolgt eine tiefgreifende genetische Diagnostik.
- Lebenstag 8–15: Anhand des genetischen Befundes und nach der Untersuchung auf Adenovirus-Antikörper wird bei der Krankenkasse die Kostenübernahme beantragt.
- Lebenstag 12–18: Nach der Bewilligung durch die Krankenkasse wird das Medikament abgestimmt auf das Körpergewicht des Kindes in Irland bestellt. Das Kind wird in der Kinderklinik des neuromuskulären Zentrums isoliert, um Infekte oder Erkrankungen auszuschließen.
- Lebenstag 18–28: Infusion des Medikaments und klinische Überwachung des Kindes.